Atlas mapping workflow#
Rather than a stand-alone classifier, this workflow maps a new query dataset onto a reference PDAC atlas in the atlas’s own SCANVI latent space, so query cells can be visualised and annotated side-by-side with the reference.
scpdac exposes two entry points:
scpdac.tl.extend_atlas— performs scArches surgery on the packaged SCANVI model, fine-tuning it on your query (which may introduce entirely new batches), then returns an expanded atlas containing both reference and query cells in a shared embedding — while retaining all of your query metadata.scpdac.tl.embed_and_predict— the lightweight, no-surgery path: it embeds the query with the frozen reference model and transfers labels, without modifying the atlas.
In this tutorial we run both on the Chen et al. 2025 cohort and compare the resulting UMAPs.
Bring your own data. The download cells below exist only for the tutorial — skip them and start from Section 2 with your own
AnnData.
Setup#
We silence third-party warnings for a cleaner notebook, then import the
single-cell stack, scpdac, and a fast approximate-nearest-neighbour transformer
for the UMAP graph.
import warnings
warnings.filterwarnings("ignore")
import pandas as pd
import anndata as ad
import scpdac
import scanpy as sc
from sklearn_ann.kneighbors.annoy import AnnoyTransformer
1. Download the query data (tutorial only)#
We use the Chen et al. 2025
PDAC cohort (GEO accession GSE278688) — 14 samples and roughly 200,000 cells —
as an example query.
Using your own dataset? Skip straight to Section 2; none of these download cells are needed.
!mkdir /home/daniele/chen_2025
!wget "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE278688&format=file&file=GSE278688%5Fsc%5Ffeatures%2Etsv%2Egz" -O /home/daniele/chen_2025/features.tsv.gz
!wget "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE278688&format=file&file=GSE278688%5Fsc%5Fbarcodes%2Etsv%2Egz" -O /home/daniele/chen_2025/barcodes.tsv.gz
!wget "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE278688&format=file&file=GSE278688%5Fsc%5Fmatrix%2Emtx%2Egz" -O /home/daniele/chen_2025/matrix.mtx.gz
!wget "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE278688&format=file&file=GSE278688%5Fsc%5Fmetadata%2Ecsv%2Egz" -O /home/daniele/chen_2025/metadata.csv.gz
--2026-06-30 10:07:51-- https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE278688&format=file&file=GSE278688%5Fsc%5Fmetadata%2Ecsv%2Egz
Resolving www.ncbi.nlm.nih.gov (www.ncbi.nlm.nih.gov)... 130.14.29.110, 2607:f220:41e:4290::110
Connecting to www.ncbi.nlm.nih.gov (www.ncbi.nlm.nih.gov)|130.14.29.110|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1166884 (1.1M) [application/octet-stream]
Saving to: ‘/home/daniele/chen_2025/metadata.csv.gz’
/home/daniele/chen_ 100%[===================>] 1.11M 1.44MB/s in 0.8s
2026-06-30 10:07:52 (1.44 MB/s) - ‘/home/daniele/chen_2025/metadata.csv.gz’ saved [1166884/1166884]
2. Load the query and the reference atlas#
The query needs raw counts and a Sample_ID batch column. We also load a
(subsampled) reference atlas to map onto — any atlas stored as zarr/h5ad with
matching raw counts works.
query = sc.read_10x_mtx("/home/daniele/chen_2025")
The published metadata uses patients as the sample identifier, so we rename it
to the Sample_ID convention used throughout scpdac.
cell_metadata = pd.read_csv("/home/daniele/chen_2025/metadata.csv.gz", index_col=0)
cell_metadata = cell_metadata.rename(
columns={
"patients": "Sample_ID",
}
)
query.obs = cell_metadata
len(query.obs.Sample_ID.unique())
14
Keep an untouched copy of the raw counts in layers["counts"] for the model to
bin and embed.
query.layers["counts"] = query.X.copy()
For a fast demo we restrict the query to two patients and map onto a 100k-cell subsample of the human atlas. Use the full atlas and query for production runs.
query = query[query.obs.Sample_ID.isin(["PA01", "PA02"])].copy()
atlas = ad.read_zarr("/data/Daniele/atlases/final_versions/Human_subsampled.zarr")
query, atlas
(AnnData object with n_obs × n_vars = 9631 × 30543
obs: 'tissue', 'Sample_ID', 'all_celltype'
var: 'gene_ids', 'feature_types'
layers: 'counts',
AnnData object with n_obs × n_vars = 100000 × 39041
obs: 'Sample_ID', 'Condition', 'Treatment', 'TreatmentType', 'TreatmentStatus', 'Tissue', 'Sex', 'Dataset', 'Technology', 'Level_1', 'Level_2', 'Level_3', 'Level_4', 'Age', 'Diabetes', 'Is_Core', 'EMT category', 'Dataset_ID', 'Cluster_Names'
var: 'n_cells', 'ensembl_id', 'start', 'end', 'chromosome', 'gene_name_adata_sc', 'highly_variable_adata_sc', 'means_adata_sc', 'dispersions_adata_sc', 'dispersions_norm_adata_sc', 'highly_variable_nbatches_adata_sc', 'highly_variable_intersection_adata_sc', 'n_cells_by_counts_adata_sc', 'mean_counts_adata_sc', 'log1p_mean_counts_adata_sc', 'pct_dropout_by_counts_adata_sc', 'total_counts_adata_sc', 'log1p_total_counts_adata_sc', 'mito_adata_sc', 'n_cells_by_counts_adata_sn', 'mean_counts_adata_sn', 'log1p_mean_counts_adata_sn', 'pct_dropout_by_counts_adata_sn', 'total_counts_adata_sn', 'log1p_total_counts_adata_sn', 'Manual_Genes', 'mt', 'ribo', 'hb'
uns: 'Condition_colors', 'Is_Core_colors', 'Level_1_colors', 'Level_2_colors', 'Level_3_colors', 'Level_4_All_colors', 'Level_4_Final_colors', 'Level_4_colors', 'UMAP_0.75', 'UMAP_0.85', '_scvi_manager_uuid', '_scvi_uuid', 'neighbors', 'umap'
obsm: 'EMT score', 'EMT_score_DL', 'Global_Leiden', 'MALAT1_lognorm', 'UMAP_0.75', 'UMAP_0.85', 'UMAP_0.95', 'X_pca', 'X_umap', '_scvi_batch', '_scvi_labels', 'batch', 'bin_edges', 'cnv_score_abs', 'empty_droplet', 'infercnv_score_malignant', 'infercnv_score_malignant_refined', 'is_outlier_total_counts', 'leiden', 'leiden_0.2', 'leiden_0.2_annotation', 'leiden_0.5', 'leiden_subcluster', 'level0_leiden_subcluster', 'log1p_n_genes_by_counts', 'log1p_total_counts', 'log1p_total_counts_mito', 'log_counts', 'mt_frac', 'n_counts', 'n_genes', 'n_genes_by_counts', 'outlier', 'pct_counts_mito', 'scANVI_cross_species', 'scANVI_emb_final', 'scanvi_L4_emb', 'scanvi_extended_atlas_emb', 'total_counts', 'total_counts_mito'
layers: 'counts', 'log_norm', 'raw'
obsp: 'connectivities', 'distances')
3. Expand the atlas with scArches surgery#
scpdac.tl.extend_atlas fine-tunes the packaged SCANVI model on the query
(max_epochs controls the surgery length), embeds both atlas and query into the
shared latent space, and concatenates them into a single expanded AnnData. The
result carries a source column (atlas / query), predicted labels in
predicted_celltype, and every original query obs column.
expanded = scpdac.tl.extend_atlas(query, atlas, species="human", max_epochs=10)
INFO File /home/daniele/Code/github_synced/scPDAC/src/scpdac/models/scanvi/human_scanvi/model.pt already
downloaded
INFO Training for 10 epochs.
Epoch 10/10: 100%|██████████| 10/10 [00:08<00:00, 1.30it/s, v_num=1, train_loss_step=1.78e+3, train_loss_epoch=1.86e+3]
Epoch 10/10: 100%|██████████| 10/10 [00:08<00:00, 1.24it/s, v_num=1, train_loss_step=1.78e+3, train_loss_epoch=1.86e+3]
expanded
AnnData object with n_obs × n_vars = 109631 × 29305
obs: 'Sample_ID', 'Condition', 'Treatment', 'TreatmentType', 'TreatmentStatus', 'Tissue', 'Sex', 'Dataset', 'Technology', 'Level_1', 'Level_2', 'Level_3', 'Level_4', 'Age', 'Diabetes', 'Is_Core', 'EMT category', 'Dataset_ID', 'Cluster_Names', 'source', 'predicted_celltype'
var: 'n_cells', 'ensembl_id', 'start', 'end', 'chromosome', 'gene_name_adata_sc', 'highly_variable_adata_sc', 'means_adata_sc', 'dispersions_adata_sc', 'dispersions_norm_adata_sc', 'highly_variable_nbatches_adata_sc', 'highly_variable_intersection_adata_sc', 'n_cells_by_counts_adata_sc', 'mean_counts_adata_sc', 'log1p_mean_counts_adata_sc', 'pct_dropout_by_counts_adata_sc', 'total_counts_adata_sc', 'log1p_total_counts_adata_sc', 'mito_adata_sc', 'n_cells_by_counts_adata_sn', 'mean_counts_adata_sn', 'log1p_mean_counts_adata_sn', 'pct_dropout_by_counts_adata_sn', 'total_counts_adata_sn', 'log1p_total_counts_adata_sn', 'Manual_Genes', 'mt', 'ribo', 'hb', 'gene_ids', 'feature_types'
obsm: 'X_scANVI_emb'
layers: 'counts'
Visualise the expanded atlas#
We build a joint UMAP directly from the shared X_scANVI_emb embedding — no PCA
needed, since the SCANVI latent space is already the integrated representation.
sc.pp.neighbors(expanded, use_rep="X_scANVI_emb", transformer=AnnoyTransformer(15))
sc.tl.umap(
expanded,
)
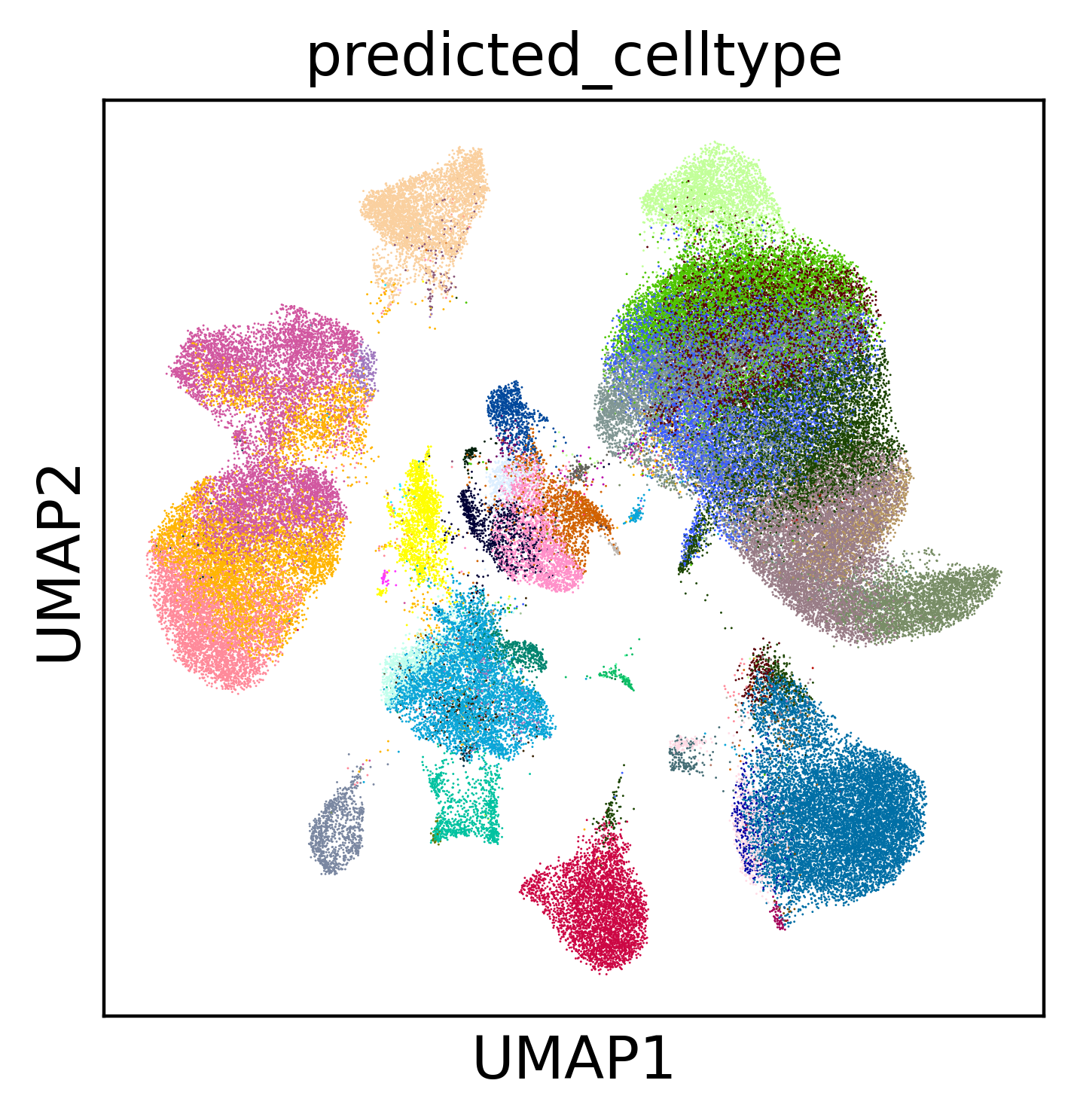
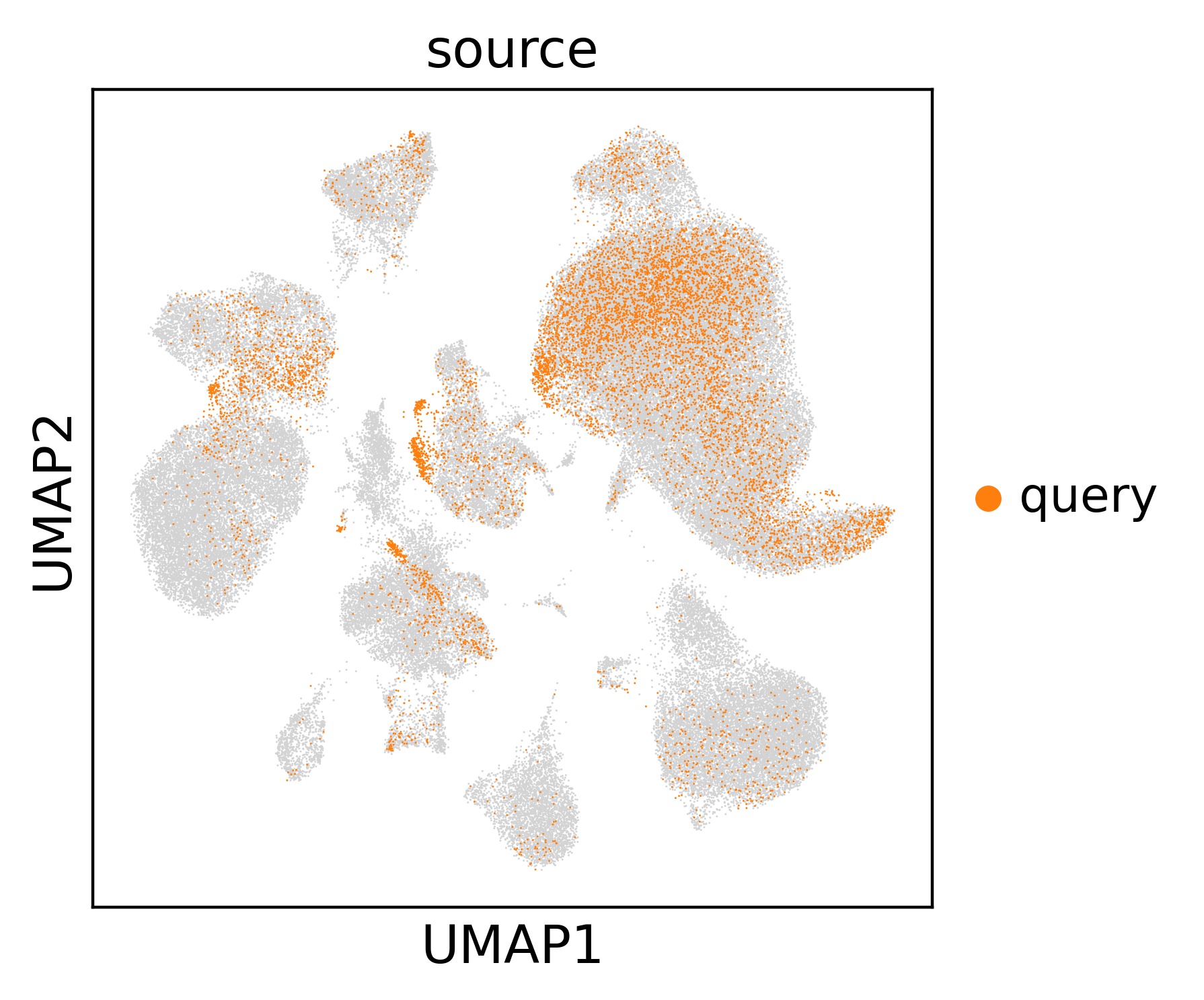
Colouring by predicted_celltype shows the transferred annotations across the
combined embedding; highlighting source == "query" shows where the new cells
land relative to the reference. Well-integrated query cells intermix with matching
reference populations rather than forming isolated islands.
sc.pl.umap(
expanded,
color="predicted_celltype",
legend_loc=None
)
sc.pl.umap(expanded, color="source", groups="query", na_in_legend=False)
expanded
AnnData object with n_obs × n_vars = 109631 × 29305
obs: 'Sample_ID', 'Condition', 'Treatment', 'TreatmentType', 'TreatmentStatus', 'Tissue', 'Sex', 'Dataset', 'Technology', 'Level_1', 'Level_2', 'Level_3', 'Level_4', 'Age', 'Diabetes', 'Is_Core', 'EMT category', 'Dataset_ID', 'Cluster_Names', 'source', 'predicted_celltype'
var: 'n_cells', 'ensembl_id', 'start', 'end', 'chromosome', 'gene_name_adata_sc', 'highly_variable_adata_sc', 'means_adata_sc', 'dispersions_adata_sc', 'dispersions_norm_adata_sc', 'highly_variable_nbatches_adata_sc', 'highly_variable_intersection_adata_sc', 'n_cells_by_counts_adata_sc', 'mean_counts_adata_sc', 'log1p_mean_counts_adata_sc', 'pct_dropout_by_counts_adata_sc', 'total_counts_adata_sc', 'log1p_total_counts_adata_sc', 'mito_adata_sc', 'n_cells_by_counts_adata_sn', 'mean_counts_adata_sn', 'log1p_mean_counts_adata_sn', 'pct_dropout_by_counts_adata_sn', 'total_counts_adata_sn', 'log1p_total_counts_adata_sn', 'Manual_Genes', 'mt', 'ribo', 'hb', 'gene_ids', 'feature_types'
uns: 'neighbors', 'umap', 'predicted_celltype_colors', 'source_colors'
obsm: 'X_scANVI_emb', 'X_umap'
layers: 'counts'
obsp: 'distances', 'connectivities'
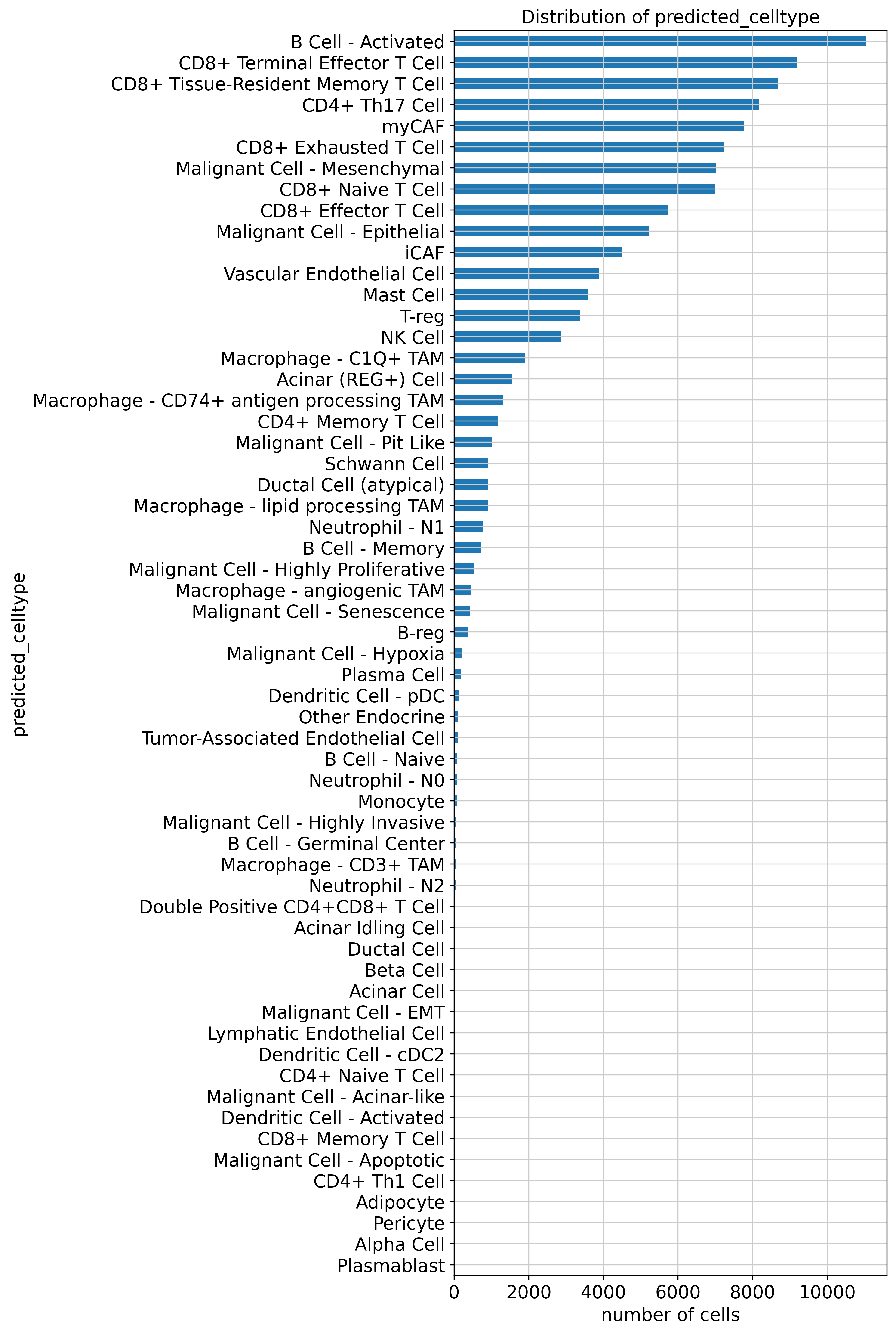
scpdac.pl.plot_label_distribution(expanded, key="predicted_celltype")
<Axes: title={'center': 'Distribution of predicted_celltype'}, xlabel='number of cells', ylabel='predicted_celltype'>
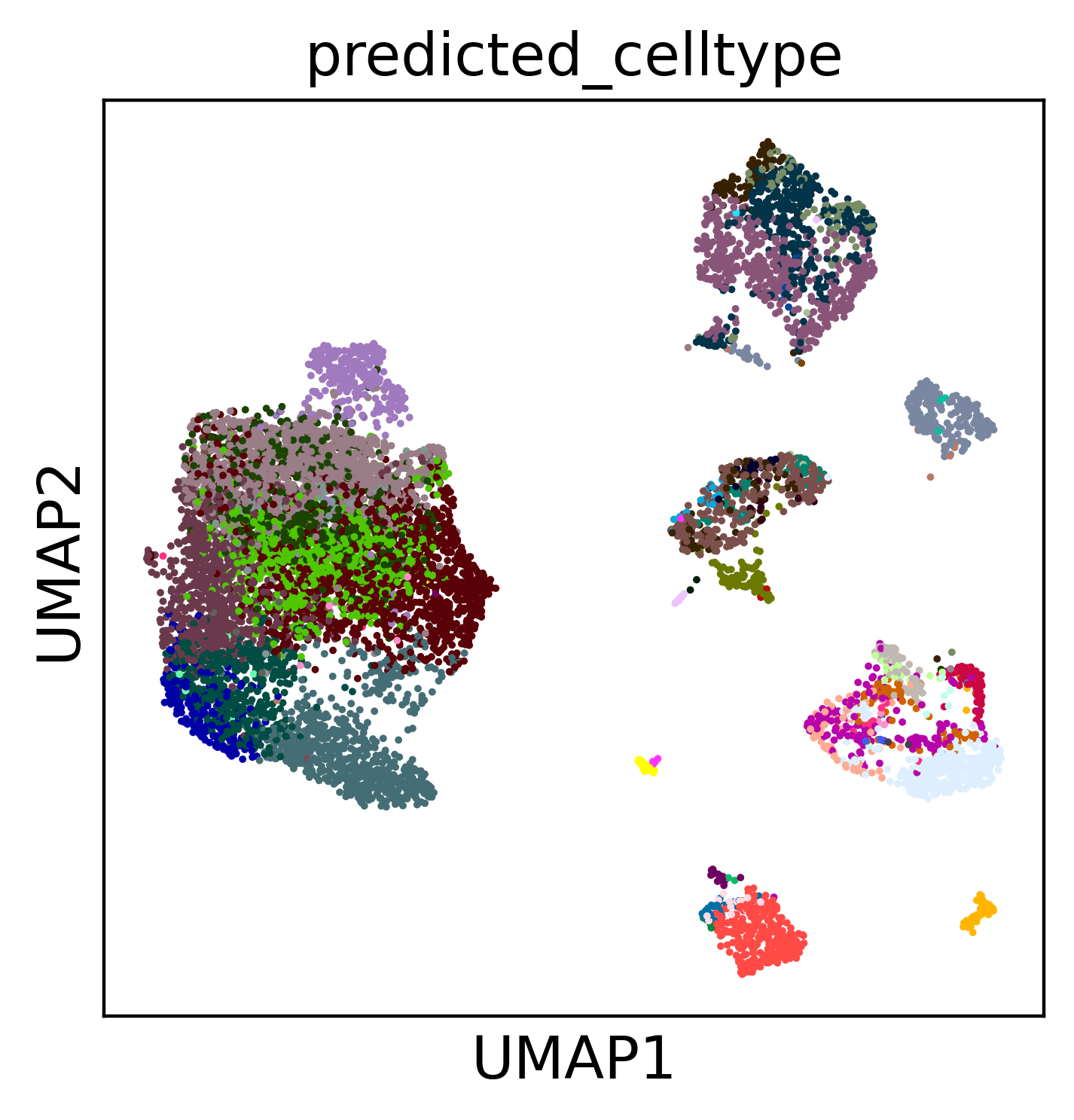
4. Embed-only: transfer labels without surgery#
If you do not want to compare your data against the rest of the atlas, you can simply use scpdac.tl.embed_and_predict which embeds the query with the frozen model and transfers labels in one step — faster, and it leaves the atlas
untouched. It writes the latent embedding to obsm["X_scANVI_emb"] and
predictions to obs["predicted_celltype"].
query = scpdac.tl.embed_and_predict(query, "human")
INFO File /home/daniele/Code/github_synced/scPDAC/src/scpdac/models/scanvi/human_scanvi/model.pt already
downloaded
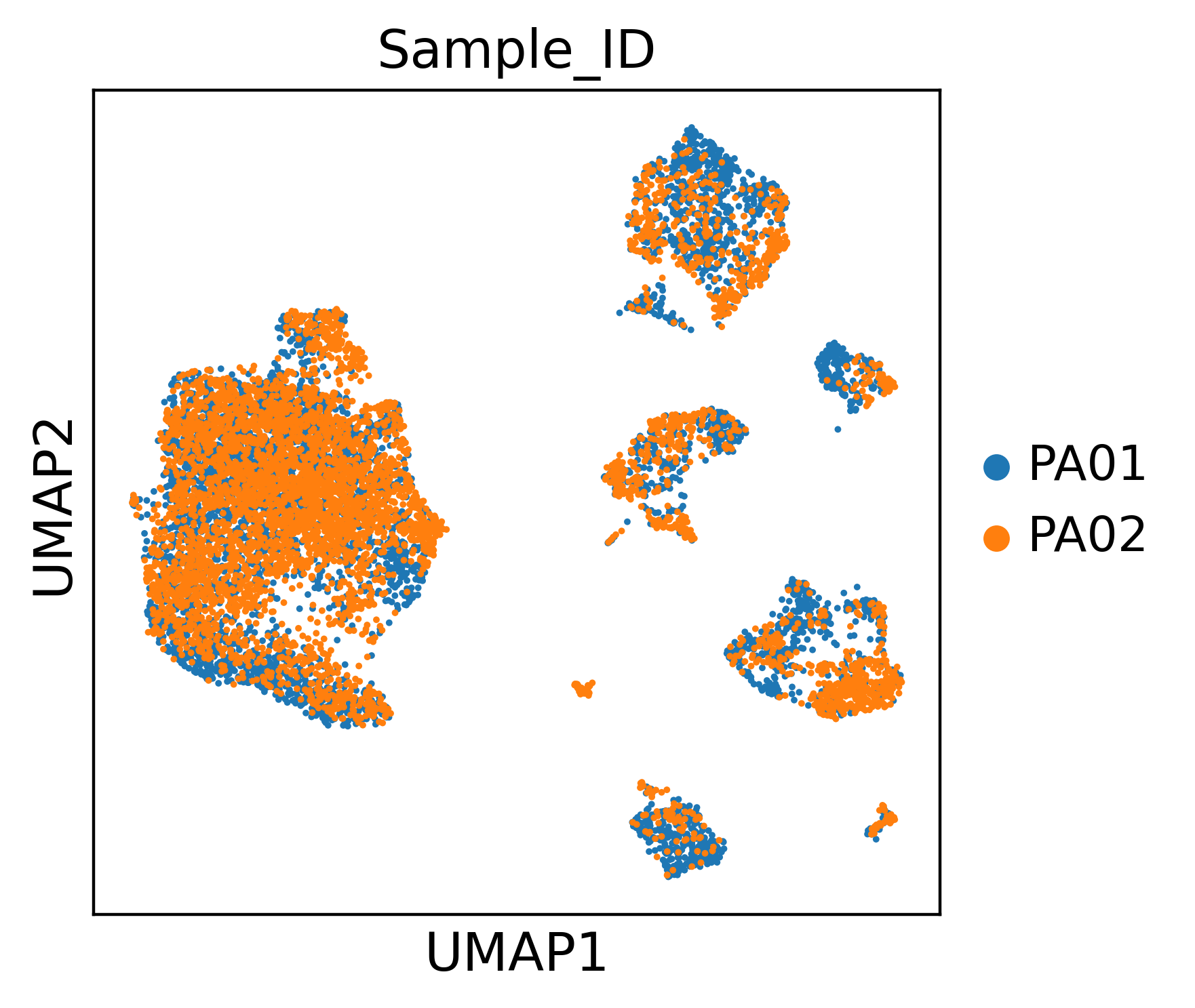
Again we compute a UMAP from the embedding and colour by the predicted cell types and by sample, to confirm the query integrates cleanly on its own.
sc.pp.neighbors(query, use_rep="X_scANVI_emb", transformer=AnnoyTransformer(15))
sc.tl.umap(
query,
)
sc.pl.umap(
query,
color="predicted_celltype",
legend_loc=None
)
sc.pl.umap(
query,
color="Sample_ID",
)